Materials Studio教程2025:从入门到精通,一篇带你玩转材料模拟的终极指南
** 还在为Materials Studio复杂的功能而头疼?本文是一份为你精心准备的2025年最新Materials Studio教程,无论你是零基础的小白,还是希望提升技能的进阶用户,这份指南都将带你从软件安装、基础操作开始,逐步深入到晶体构建、性质计算和结果分析的核心环节,我们将提供详细的图文步骤、实用技巧和常见问题解答,助你高效掌握这款强大的材料模拟软件,让你的科研工作事半功倍。

引言:为什么选择Materials Studio?它究竟能为你做什么?
作为一名程序员,我深知“工欲善其事,必先利其器”的道理,在材料科学、化学、物理和工程领域,Materials Studio(简称MS)无疑是一把锋利的“瑞士军刀”,它是由美国Accelrys公司(现为BIOVIA公司)开发的一款领先的材料建模与模拟软件套件。
它能帮你做什么?
- 预测材料性能: 在实验室合成之前,通过计算机模拟预测材料的力学、电学、光学、热学等关键性能,大大缩短研发周期,降低试错成本。
- 理解微观机理: 从原子、分子层面理解化学反应、相变、扩散等微观过程的机理,为实验提供理论指导。
- 结构优化设计: 针对你的特定需求(如催化剂、电池材料、药物分子),进行分子或晶体结构的优化设计,寻找最佳构型。
MS功能强大,但也伴随着陡峭的学习曲线,很多人面对其繁多的模块和复杂的界面望而却步,别担心,这份Materials Studio教程就是你的“导航地图”,我们将用程序员式的逻辑思维,一步步拆解它,让你轻松上手。
Materials Studio教程第一部分:新手入门——你的第一步
在开始模拟之前,确保你已经完成了准备工作。

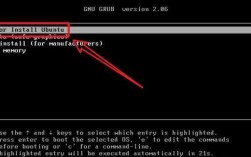
软件安装与激活
- 获取版本: 建议从学校或研究机构的正版渠道获取,常用的有MS 8.0、9.0及更高版本,对于个人学习,可寻找社区版或试用版。
- 安装步骤:
- 关闭杀毒软件和防火墙(安装完成后务必重新开启)。
- 以管理员身份运行安装程序。
- 按照向导提示进行安装,选择“完整安装”以包含所有模块。
- 安装完成后,使用提供的许可文件(
.lic)进行配置,通常需要修改C:\Program Files (x86)\BIOVIA\MaterialsStudioXX\etc\目录下的hosts文件,将localhost指向0.0.1,然后运行LicenseServer激活。
- 常见问题: 激活失败通常是
hosts文件或防火墙设置问题。程序员小贴士: 善用文本编辑器(如VS Code)修改hosts文件,检查语法是否正确。
熟悉工作界面
启动MS,你会看到一个类似“项目管理器”和多个“模块”的界面。
- Project Explorer(项目管理器): 位于左侧,是所有文件和操作的中心,在这里创建、打开、管理你的项目。
- Modules(模块): 位于顶部,按功能分类(如
Build、Discover、Refine等),每个模块都包含一系列工具。 - Visualization(可视化窗口): 中间最大的区域,用于显示你的分子、晶体模型,这是你与材料“对话”的主要窗口。
- Scripting(脚本): 位于底部,是高级用户的利器,你可以在这里编写Python或Delphi脚本,实现自动化操作和复杂计算。程序员必看: 学习脚本是提升效率的关键!
- Properties(属性面板): 右侧,显示选中对象的详细信息,如原子坐标、键长、键角等。
新手任务: 尝试在Project Explorer中新建一个项目,然后在Build模块下,使用Sketch工具画一个简单的H₂O分子,并在Visualization窗口中旋转、缩放它。
Materials Studio教程第二部分:核心实战——从原子到性质
这是教程的核心部分,我们将通过一个完整的实例,带你走完一次材料模拟的全流程。
案例:构建金红石结构TiO₂并计算其能带结构
目标: 学习如何构建晶体模型,并使用CASTEP模块计算其电子结构。

步骤1:构建晶体模型
- 新建项目: 在
Project Explorer中,右键新建一个项目,命名为TiO2_Rutile。 - 启动工具: 在
Build模块中,双击Crystallizer工具。 - 设置参数:
- 在
Space Group(空间群)中输入金红石TiO₂的空间群编号:P4₂/mnm。 - 在
Lattice Parameters(晶格参数)中,输入实验值或文献值:- a = 4.593 Å
- c = 2.958 Å
- 点击
Build按钮。
- 在
- 查看结果:
Visualization窗口中会出现一个TiO₂晶胞,你可以通过Build->Symmetry->Supercell工具构建超胞,以便更清晰地观察结构。
步骤2:结构优化(Geometry Optimization)
为了得到能量最低、最稳定的结构,我们必须先进行结构优化。
- 启动模块: 在
Discover模块中,双击CASTEP。 - 设置任务: 在右侧
Setup面板中,将Task(任务)设置为Geometry Optimization(几何优化)。 - 选择赝势: 在
Pseudopotentials(赝势)选项卡中,选择Ti和O元素的赝势,通常选择OTF(On-the-fly)赝势,它会自动生成,非常方便。 - 设置精度: 在
Quality(质量)选项卡中,选择Medium(中等)或Fine(精细),精度越高,计算时间越长,结果越准,新手建议从Medium开始。 - 运行计算: 点击工具栏上的绿色的
Run按钮,计算可能需要几分钟到几十分钟不等,请耐心等待,计算进度会显示在Jobs窗口中。
步骤3:分析优化后的结构
计算完成后,你会得到一个.castep文件。
- 打开结果: 在
Project Explorer中,双击TiO2_Rutile.castep文件。 - 查看信息: 软件会自动打开结果查看器,你可以查看
Geometry Optimization标签页下的最终能量、晶格常数变化等,确认结构是否已收敛。 - 可视化: 回到
Visualization窗口,你会发现模型已经更新为优化后的结构。
步骤4:计算能带结构
结构优化后,我们进行电子性质计算。
- 复制任务: 在
Project Explorer中,右键点击之前的CASTEP计算任务,选择Copy,我们将基于已优化的结构进行新计算。 - 修改设置: 双击新的
CASTEP任务。- 将
Task修改为Electronic(电子结构)。 - 在
Properties(性质)选项卡中,勾选Band Structure(能带结构)。 - 在
Parameters(参数)选项卡中,设置k-point路径,对于金红石结构,一个典型的路径是G -> X -> M -> G -> R -> Z,你可以使用k-point Path Generator工具来生成。
- 将
- 运行计算: 再次点击
Run按钮,这次计算会比几何优化更快。
步骤5:解读能带图
计算完成后,双击新的.castep文件,在Band Structure标签页下,你将看到美丽的能带图。
- 横坐标 (k-path): 表示布里渊区中的高对称点路径。
- 纵坐标 (Energy / eV): 表示电子能量。
- 费米能级 (Fermi Level): 图中通常有一条水平虚线表示。关键判断: 如果价带顶(最高能带)和导带底(最低能带)都穿过费米能级,材料是金属;如果价带顶在费米能级以下,导带底在费米能级以上,且两者之间存在能量差,这个差值就是带隙,材料是半导体或绝缘体。